
David Van Vactor (HMS, Dept. of Cell Biology)
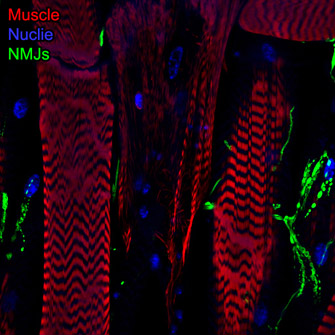
Spinal Muscular Atrophy (SMA), the leading cause of genetically linked infant mortality, is an autosomal, recessive neurodegenerative disease that results in degeneration of spinal cord motoneurons and muscle atrophy, and is the result of the loss of survival motor neuron 1 (SMN1) gene activity. We aim to identify genetic factors that protect against degenerative aspects of SMA phenotypes, including morphological and functional defects in the neuromuscular junction (NMJ) and progressive muscle loss. Using Drosophila, we have developed models and experimental tools that allow us explore questions related to intermediate late stages of disease progression. We identified genetically tractable Smn-dependent fly models that recapitulate the degenerative effects of SMA. These models provide an opportunity to define novel genes and/or pathways that can protect against these late-stage features by modulating SMA phenotypes. As we have performed a genetic modifier screen for genes that confer protection against Smnreduction (Chang, et al.; Sen, et al.), we are in a unique position to dissect genes andpathways that have the ability to ameliorate progressive/late-stage NMJ and muscle defects. Moreover, the identification of effective modifiers of SMN function may very well define useful biomarkers to evaluate the efficacy of a drug (e.g., Nusinersen or “Spinraza”) or may define secondary targets for which modulation may positively affect the therapeutic window of Nusinersen.
The genetic screens we carried out identified elements of the Fibroblast Growth Factor (FGF), Transforming Growth Factor beta (TGF-?), Wingless/WNT pathways as SMN modifiers. We are interested in further pursuing the mechanisms behind these interactions, especially as all three pathways have been implicated in synaptic development and maintenance in vertebrates.