Neurodegenerative Disease Research
We are using Drosophilamodels to study Amyotrophic Lateral Sclerosis (ALS). ALS, also known as Lou Gehrig’s Disease, is a devastating, and invariably fatal, neurodegenerative disorder that strikes during adulthood and causes rapid, progressive loss of motor neurons. Existing fruit fly models overexpressing disease-causing alleles of genes such as Tdp43, Fus, and c9orf72(G4C2)n recapitulate many aspects of ALS in humans, including neuronal degeneration, reduced lifespan, and locomotor defects. However, expression of disease genes and the appearance of their related phenotypes in these models begins during development or immediately thereafter, in contrast to the human disease where the average age of diagnosis is 55. Therefore, in order to generate fly models that more closely mimic human ALS, we are testing strategies that block the expression of disease genes during development, thus creating a true adult-onset model of the disease. We are examining the functional effects of ALS gene expression in adult flies by assessing effects on lifespan, locomotor activity, and motor neuron morphology. We are also testing the effects of gain- and loss-of-function for candidate ALS modifier genes (which were identified in a previous genetic screen conducted in the Artavanis-Tsakonas lab) in these adult-onset models.
Tau and A?42 modifiers
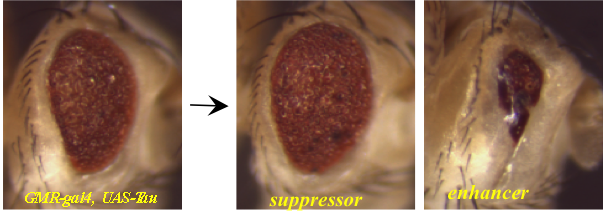
Alzheimer’s Disease (AD) is the most common cause of dementia in the aging population and may affect up to one-third of individuals over 85. Although a number of genes have been associated both genetically and functionally with AD, there is little doubt that many more unidentified genes contribute to AD risk, pathology, and progression. In order to identify some of these factors, we are performing large-scale genetic screens in the fly to identify genes that interact with the AD-related genes Tau and A?42 to modify a neurodegeneration phenotype in the fly eye. For each of these screens, we test approximately 16,000 individual mutant fly lines. Potential modifiers identified in these screens will be further validated with additional functional assays in both fly and mammalian models. These genes may indicate novel pathways that interact with Tau and A?42 during AD progression, and may be potential therapeutic targets.
ApoE4 Modifiers
The E4 variant of the Apolipoprotein E gene (ApoE4) is the single greatest genetic risk factor for developing Alzheimer’s Disease (AD); however, the functional role of ApoE4 protein in AD remains largely opaque. We are interested in identifying additional genes that may interact with ApoE4 to affect AD phenotypes, in order to elucidate the mechanisms through which ApoE4 acts to affect both AD pathogenesis and normal biology. We are therefore conducting a suppressor screen using the Exelixis collection of gain- and loss-of-function fly mutants to identify genes that suppress a strong ApoE4 lethality effect in the fly. Modifiers identified in this screen will be further studied in the context of ApoE4 using a variety of genetic, biochemical, and cell biological experiments.